XClone tutorials on TNBC1 scRNA-seq
A triple-negative breast cancer (TNBC) sample that was assayed by droplet-based scRNA-seq (10x Genomics), published by CopyKAT with dataset ID as TNBC1. The aligned reads in BAM format and the called cell list were directly downloaded from GEO GSE148673. As reported by the original study using CopyKAT, three clusters of cells were identified from the transcriptome, consisting of 300 normal cells and 797 tumour cells from two distinct CNV clones.
Download the Jupyter Notebook by clicking on the Download TNBC1 scRNA-seq demo Notebook.
Introduction
This tutorial covers how to use XClone for scRNA-seq CNV analysis in RDR module, BAF module and Combine module independently.
Author: Rongting Huang
Date: 2023-04-03
The data used in this tutorial is available in XClone package and can be download from xclonedata.
The data contains:
Single-cell RNA-seq read count data for XClone RDR module and BAF module.
Requirements
To follow this tutorial, you will need:
Python 3.7.
The following Python packages: XClone and its dependent packages, e.g., anndata, numpy, scipy, pandas, scanpy.
In this tutorial, we covered how to use XClone for scRNA CNV analysis. If you have any questions or comments, please feel free to contact the author.
Load packages
[1]:
%load_ext autoreload
%autoreload 2
import xclone
import anndata as an
import pandas as pd
import numpy as np
import scipy
print("scipy", scipy.__version__)
xclone.pp.efficiency_preview()
(Running XClone 0.3.4)
2023-04-03 10:46:28
scipy 1.7.0
[XClone efficiency] multiprocessing cpu total count in your device 112
[2]:
dataset_name = "TNBC1_scRNA"
## output results dir
outdir = "/storage/xxxx/users/xxxx/xclone/tutorials/"
To specify the name of your dataset and the output directory where the results will be saved, you can set the dataset_name and outdir parameters before you start using any module in XClone. Replace “BCH869_scRNA” with a name of your choice to identify your dataset, and "/storage/xxxx/users/xxxx/xclone/tutorials/" with the path to the directory where you want to save the output files.
Load dataset
For TNBC1 dataset, users can easily load the raw read count matrices for RDR module and BAF module, respectively by xclone.data.tnbc1_rdr() and xclone.data.tnbc1_baf().
Users can load the matricesin anndata object in Python, it typically contains several matrices or tables of data, each with its own set of row and/or column annotations. The obs attribute typically contains annotation information for each cell in the dataset, and the var attribute typically contains annotation information for each gene in the dataset.
obs attribute used in XClone:
cluster: Cluster identifier for each cell by combining expression with BAF analysis. We annotated the cell clusters into two subclones Clone 1 (Clone A), Clone 2 (Clone B) and Normal cells.
cluster.pred: Normal and tumor cells, 'N' and 'T' here, from the above mentioned annotation.
copykat: Cluster identifier for each cell by CopyKAT publication.
copykat.pred: Normal and tumor cells identified by copyKAT publication.
Additional cell-level annotations can be merged in obs by users.
var attribute used in XClone:
GeneName: Gene Names for each gene in the dataset.
GeneID: Identifier or accession number of each gene in the dataset(if available).
chr: Chromosome location of each gene.
start: Start position of each gene on its chromosome.
stop: End position of each gene on its chromosome.
arm: Chromosome arm information for each gene.
chr_arm: Combined Chromosome and arm information for each gene.
band: Chromosome band information for each gene (if available)..
Additional gene-level annotations can be merged in var by users.
For RDR module, there is raw_expr layer in the anndata, and for BAF module, there are two layers AD and DP, initially.
[3]:
RDR_adata = xclone.data.tnbc1_rdr()
BAF_adata = xclone.data.tnbc1_baf()
RDR module
By calling the config method xclone.XCloneConfig(), users can create an instance of the XCloneConfig class in the module of “RDR” to set data-specific configures for RDR module.
In RDR module, specify a cell_anno_key with a cell annotation key used in your dataset, and ref_celltype as the reference cell type.
Set the output directory by specifying outdir.
XClone can defaultly detect and remove celltype-specific marker genes when users specify cell annotation key for marker_group_anno_key and N top marker genes in each cell group to be removed by specify the number in top_n_marker, \(N=15\) by default. If marker_group_anno_key is not defined, cell_anno_key will be used by default.
Users can decide to plot the main figures in each module or not by setting xclone_plot, True or False. If True, can also select cell annotation term plot_cell_anno_key for plotting cells by clusters.
XClone has default start probability for start_prob ([0.1, 0.8, 0.1] for copy loss, copy neutral and copy gain) in 10x scRNA-seq dataset.
Before running the RDR moudle, users can use xconfig.display() to print all configurations used in RDR module for previewing and logging.
Users can run the analysis on your input data by calling the run_RDR method by specifying the RDR_adata and config_file. If no custmoised config file is used, XClone will load the default config file.
Here shows the example of BCH869 RDR module.
[4]:
xconfig = xclone.XCloneConfig(dataset_name = dataset_name, module = "RDR")
xconfig.set_figure_params(xclone= True, fontsize = 18)
xconfig.outdir = outdir
xconfig.cell_anno_key = "cluster.pred"
xconfig.ref_celltype = "N"
xconfig.marker_group_anno_key = "cluster.pred"
xconfig.xclone_plot= True
xconfig.plot_cell_anno_key = "cluster"
xconfig.display()
RDR_Xdata = xclone.model.run_RDR(RDR_adata,
config_file = xconfig)
RDR
Configurations:
HMM_brk chr_arm
KNN_neighbors 10
WMA_smooth_key chr_arm
WMA_window_size 40
_file_format_data h5ad
_file_format_figs pdf
_frameon True
_outdir /storage/yhhuang/users/rthuang/xclone/tutorials
_plot_suffix
_start 1680518803.4498727
_vector_friendly True
cell_anno_key cluster.pred
dataset_name TNBC1_scRNA
dispersion_celltype None
exclude_XY False
file_format_data h5ad
file_format_figs pdf
filter_ref_ave 0.5
fit_GLM_libratio False
gene_exp_group 1
gene_exp_ref_log True
guide_chr_anno_key chr_arm
guide_cnv_ratio None
guide_qt_lst [0.0001, 0.96, 0.99]
marker_group_anno_key cluster.pred
max_iter 2
min_iter 1
module RDR
outdir /storage/yhhuang/users/rthuang/xclone/tutorials
plot_cell_anno_key cluster
plot_suffix
rdr_plot_vmax 0.7
rdr_plot_vmin -0.7
ref_celltype N
remove_guide_XY False
remove_marker True
select_normal_chr_num 4
set_figtitle True
set_smartseq False
smart_transform False
start_prob [0.1 0.8 0.1]
top_n_marker 15
trans_prob [[9.99998e-01 1.00000e-06 1.00000e-06]
[1.00000e-06 9.99998e-01 1.00000e-06]
[1.00000e-06 1.00000e-06 9.99998e-01]]
trans_t 1e-06
warninig_ignore True
xclone_plot True
[XClone RDR module running]************************
[XClone data preprocessing] check RDR raw dataset value: success
Keep valid cells: Filter out 0 cells / 1097 total cells, remain 1097 valid cells with annotation
[XClone data preprocessing] check RDR cell annotation: success
[XClone-RDR preprocessing] Filter out 16315 genes / 33472 total genes, remain 17157 genes
[XClone data preprocessing] detect RDR genes: done
[XClone-RDR preprocessing] Filter out 10724 genes / 17157 total genes, remain 6433 genes
Trying to set attribute `.var` of view, copying.
[XClone] use marker genes provided by users:
['AC036214.3' 'AC093484.3' 'AL365205.1' 'ATP1A1' 'B2M' 'CD24' 'CRYAB'
'CST3' 'CTSB' 'CYBA' 'DPYD' 'EMP3' 'EPCAM' 'GSTO1' 'H3F3A' 'HLA-A' 'HNMT'
'HSP90AB1' 'KRT7' 'MRPL14' 'POLR1C' 'RAB31' 'RPL28' 'TM4SF1' 'TMSB4X'
'TOMM6' 'TPD52' 'UBB' 'YIPF3' 'ZEB2']
filter_genes_num: 30
used_genes_num: 6403
output anndata is not sparse matrix.
Trying to set attribute `.var` of view, copying.
Trying to set attribute `.var` of view, copying.
[XClone RDR gene dispersion fitting] Time used: 1413 seconds
[XClone RDR gene-specific dispersion]: checking
max_value: 3.1872219121269994e+33
min_value: 2.967852820468968e-52
qt_0.95_value: 4.299742553561531
qt_0.05_value: 0.045671615314120464
remove no GLM results genes num: 0
remove inf dispersion genes num: 0
[XClone RDR dispersion]: clipping
[XClone RDR gene-specific dispersion]: checking
max_value: 2.920260325742404
min_value: 0.014103096362288924
qt_0.95_value: 2.920260325742404
qt_0.05_value: 0.045671615314120464
[XClone hint] RDR_base_file and bulk_file saved in /storage/yhhuang/users/rthuang/xclone/tutorials/data/.
make WMA connectivities matrix, saved in varp[WMA_connect].
[XClone] RDR CNV states chrs guiding(copy loss, copy neutral, copy gain): ['19q', '3p', '8q']
CNV loss: 0.6554608169760507
CNV neutral: 1.2388721424569358
CNV gain: 2.2130883651915747
[XClone] RDR CNV states ratio guiding(copy loss, copy neutral, copy gain): [0.65546082 1.23887214 2.21308837]
expression_brk [-0.6898304 5.8300295]
[XClone] CNV_optimazation iteration: 1
Cell level: no filtering emm_prob
Gene level: filter nan emm_prob
[XClone HMM smoothing] Time used: 61 seconds
[XClone] CNV_optimazation iteration: 2
[XClone] fit CNV ratio
26 2.920260
27 2.920260
38 2.920260
39 0.223667
43 0.175076
...
33348 0.779464
33350 0.424299
33353 0.115477
33355 0.152878
33357 0.159337
Name: dispersion_capped, Length: 6402, dtype: float64
[XClone] GLM success:
[0. 0. 0. ... 5. 9. 3.] [1. 1. 1. ... 1. 1. 1.] [0.4345959 0.5970708 0.71850723 ... 2.0021474 2.9733129 2.491469 ] [1. 1. 0. ... 0. 0. 0.]
[XClone] GLM success:
[0. 0. 0. ... 5. 9. 3.] [1. 1. 1. ... 1. 1. 1.] [0.4345959 0.5970708 0.71850723 ... 2.0021474 2.9733129 2.491469 ] [0. 0. 1. ... 0. 1. 1.]
[XClone] GLM success:
[0. 0. 0. ... 5. 9. 3.] [1. 1. 1. ... 1. 1. 1.] [0.4345959 0.5970708 0.71850723 ... 2.0021474 2.9733129 2.491469 ] [0. 0. 0. ... 1. 0. 0.]
Time used 8 seconds
Cell level: no filtering emm_prob
Gene level: filter nan emm_prob
[XClone HMM smoothing] Time used: 63 seconds
[XClone] Warning: Lower bound decreases!
Step 1, loglik decrease from -7.61e+06 to -7.61e+06
iteration_end_round: 2
Logliklihood: [-7609360.65300615 -7613910.98611593]
CNV_ratio: {'0': array([[0.65546082, 1.23887214, 2.21308837]]), '1': array([[0.57053172, 1.17758586, 2.7366992 ]])}
Time used 213 seconds
[XClone hint] RDR_final_file saved in /storage/yhhuang/users/rthuang/xclone/tutorials/data/.
[XClone plotting]
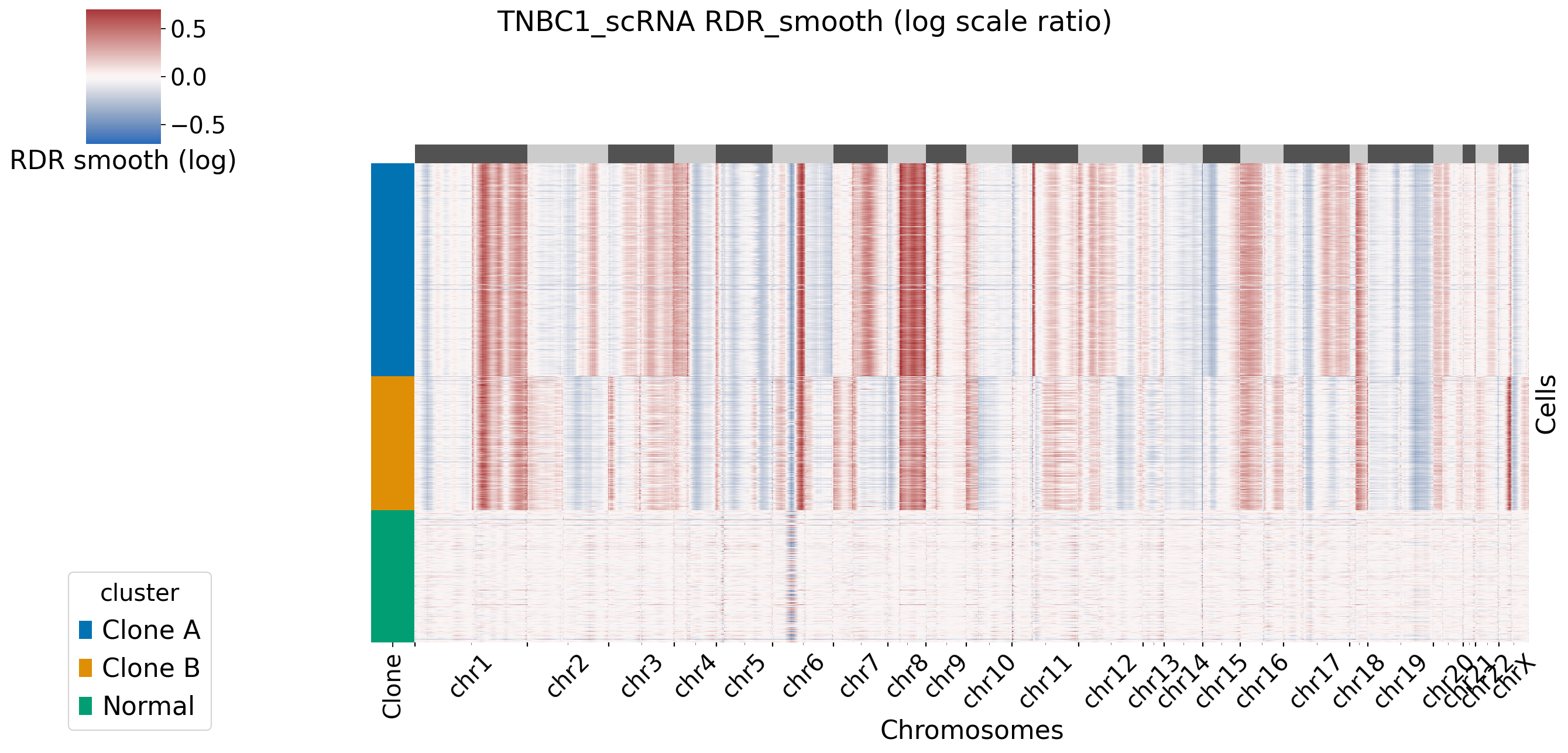
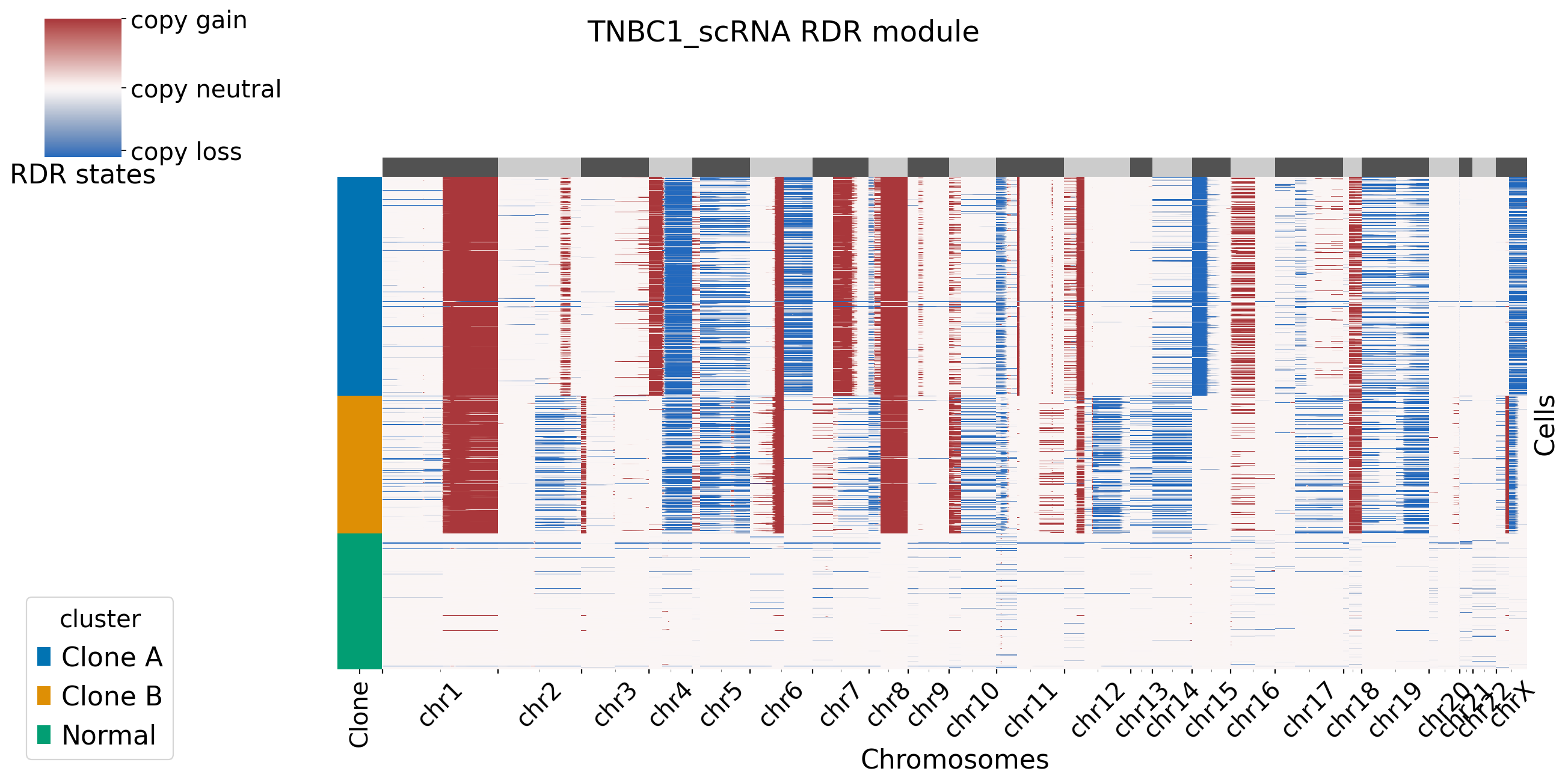
[5]:
RDR_Xdata
[5]:
AnnData object with n_obs × n_vars = 1097 × 6402
obs: 'copykat.pred', 'cluster.pred', 'cluster', 'mit_clone_id', 'confident', 'tumor', 'copykat', 'counts_ratio'
var: 'GeneName', 'GeneID', 'chr', 'start', 'stop', 'arm', 'chr_arm', 'band', 'ref_avg', 'dispersion', 'gene_dispersion_bse', 'dispersion_capped'
uns: 'data_mode', 'data_notes', 'genome_mode', 'log', 'rank_marker_genes', 'fit_dispersion_removed_genes', 'dispersion_base_celltype', 'pca', 'neighbors', 'chr_dict', 'guide_CNV_chrs_use_layers', 'guide_CNV_chrs_use_anno_key', 'ref_log_expression_brk', 'group_genes', 'CNV_ratio', 'Logliklihood'
obsm: 'X_pca'
varm: 'PCs'
layers: 'raw_expr', 'raw_ratio', 'ref_normalized', 'expected', 'WMA_smoothed', 'RDR_smooth', 'emm_prob_log', 'emm_prob_log_noHMM', 'emm_prob_noHMM', 'posterior_mtx', 'posterior_mtx_log'
obsp: 'distances', 'connectivities'
[6]:
RDR_Xdata.layers["posterior_mtx"].shape
[6]:
(1097, 6402, 3)
[7]:
RDR_Xdata.layers["posterior_mtx"].argmax(axis=-1).shape
[7]:
(1097, 6402)
Users can load the step by step results(AnnData) stored in outdir/data.
For the final RDR module CNV detection results, load the AnnData object results into memory:
import anndata
filename = outdir+"data/RDR_adata_KNN_HMM_post.h5ad"
adata = anndata.read_h5ad(filename)
Access the posterior_mtx layer:
cnv_prob = adata.layers["posterior_mtx"]
This will extract the final CNV calling results as a NumPy array.
The posterior_mtx layer contains the posterior probabilities of each cell at each genomic segment(gene scale default) having each copy number state. The rows correspond to cells and the columns correspond to genomic segments.
copy_states = cnv_prob.argmax(axis=-1)
This will give you a 2D numpy array hard assigned CNV states for each cell each gene.
For more to explore the results of XClone, pls refer to link.
BAF moudle
Similarly, by calling the config method xclone.XCloneConfig(), users can create an instance of the XCloneConfig class in the module of “BAF” to set data-specific configures for BAF module.
It is default set baf_bias_mode=1, which supports \(K=5\) BAF states: allele A bias (++), allele A bias (+), allele balance, allele B bias (+), and allele B bias (++). XClone provides \(K=3\) BAF states for comparasion (visualization). In this mode, CNV_N_components is 5. Alternatively, users can specify another mode by setting baf_bias_mode=0, which supports \(K=3\) BAF states: allele A bias,allele balance, allele B bias. In this mode, CNV_N_components is 3.
In BAF module, the start probabilitystart_prob is default set as \([0.3, 0.4, 0.3]\) and \([0.2, 0.15, 0.3, 0.15, 0.2]\) for \(K=3\) and \(K=5\), respectively. And the transition probability trans_prob for HMM smoothing is default set as \(\{t = 1e-6\), \(1-(K-1)t\}\) respectively for cross-state transition and state keeping. If users change CNV_N_components manually, should also consider the correspoding start probability and transition probability in HMM
settings.
Generaly, users need specify a cell_anno_key with a cell annotation key used in your dataset, and ref_celltype as the reference cell type.
Set the output directory by specifying outdir.
Generaly, data-specific theoractical B allele frequency is fitted from the reference cells. However, if the reference cells is limited, users can set theo_neutral_BAF=0.5 directly. Here we use default setting in TNBC1 dataset.
Users can decide to plot the main figures in each module or not by setting xclone_plot, True or False. If True, can also select cell annotation term plot_cell_anno_key for plotting cells by clusters. XClone perform the denoise strategy in BAF module and plot the denoised BAF CNV profile by default.
Before running the BAF moudle, users can use xconfig.display() to print all configurations used in RDR module for previewing and logging.
Users can run the analysis on your input data by calling the run_BAF method by specifying the BAF_adata and config_file. If no custmoised config file is used, XClone will load the default config file.
Here shows the example of BCH869 BAF module.
[8]:
xconfig = xclone.XCloneConfig(dataset_name = dataset_name, module = "BAF")
xconfig.set_figure_params(xclone= True, fontsize = 18)
xconfig.outdir = outdir
xconfig.cell_anno_key = "cluster.pred"
xconfig.ref_celltype = "N"
xconfig.xclone_plot= True
xconfig.plot_cell_anno_key = "cluster"
xconfig.display()
BAF_merge_Xdata = xclone.model.run_BAF(BAF_adata,
config_file = xconfig)
BAF
Configurations:
BAF_add None
BAF_denoise True
BAF_denoise_GMM_comp 2
BAF_denoise_GMM_detection True
BAF_denoise_cellprop_cutoff 0.05
CNV_N_components 5
HMM_brk chr_arm
KNN_neighbors 10
RDR_file None
WMA_smooth_key chr_arm
WMA_window_size 101
_file_format_data h5ad
_file_format_figs pdf
_frameon True
_outdir /storage/yhhuang/users/rthuang/xclone/tutorials
_plot_suffix
_start 1680520658.023401
_vector_friendly True
baf_bias_mode 1
bin_nproc 20
cell_anno_key cluster.pred
concentration 100
concentration_lower 20
concentration_upper 100
dataset_name TNBC1_scRNA
exclude_XY False
extreme_count_cap False
feature_mode GENE
file_format_data h5ad
file_format_figs pdf
gene_specific_concentration False
guide_theo_CNV_states None
module BAF
outdir /storage/yhhuang/users/rthuang/xclone/tutorials
phasing_len 100
phasing_region_key chr
plot_cell_anno_key cluster
plot_suffix
ref_BAF_clip False
ref_celltype N
remove_guide_XY False
remove_marker_genes True
set_figtitle True
set_smartseq False
start_prob [0.2 0.15 0.3 0.15 0.2 ]
theo_neutral_BAF None
trans_prob [[9.99996e-01 1.00000e-06 1.00000e-06 1.00000e-06 1.00000e-06]
[1.00000e-06 9.99996e-01 1.00000e-06 1.00000e-06 1.00000e-06]
[1.00000e-06 1.00000e-06 9.99996e-01 1.00000e-06 1.00000e-06]
[1.00000e-06 1.00000e-06 1.00000e-06 9.99996e-01 1.00000e-06]
[1.00000e-06 1.00000e-06 1.00000e-06 1.00000e-06 9.99996e-01]]
trans_t 1e-06
warninig_ignore True
xclone_plot True
[XClone BAF module running]************************
[XClone data preprocessing] check BAF raw dataset value: success
Keep valid cells: Filter out 0 cells / 1097 total cells, remain 1097 valid cells with annotation
[XClone data preprocessing] check BAF cell annotation: success
[XClone data checking]: RDR and BAF in same cell order.
[XClone-data removing]:
Filter out 30 genes / 33472 total genes, remain 33442 regions
[XClone-Local_phasing] time_used: 76.63seconds
[XClone-Global_phasing] time_used: 3.32seconds
make WMA connectivities matrix, saved in varp[WMA_connect].
WMA_connect exists for direct use.
... storing 'bin_stop_arm' as categorical
... storing 'bin_stop_chr_arm' as categorical
... storing 'bin_stop_band' as categorical
[XClone hint] BAF_base_file and merged_file saved in /storage/yhhuang/users/rthuang/xclone/tutorials/data/.
[XClone get_CNV_states] time_used: 303.31seconds
states used: [0.24059262 0.3962636 0.50714286 0.63970433 0.79573162]
.....
[XClone] specific Center states used.
[XClone]: validated probability, all finite.
cal emm prob time 7 seconds
normalize the input emm_prob_log
normalized emm_prob_log
generate new layer key value: bin_phased_BAF_specific_center_emm_prob_log_KNN
[BAF smoothing] time_used: 1.93seconds
Cell level: no filtering emm_prob
Gene level: no filtering emm_prob
[XClone] multiprocessing for each brk item
nproc: 80
[XClone HMM smoothing] Time used: 11 seconds
[XClone get_CNV_states] time_used: 204.47seconds
states used: [0.28692468 0.50714286 0.72361922]
.....
[XClone] specific Center states used.
[XClone]: validated probability, all finite.
cal emm prob time 1 seconds
normalize the input emm_prob_log
normalized emm_prob_log
generate new layer key value: correct_emm_prob_log_KNN
[BAF smoothing] time_used: 1.04seconds
Cell level: no filtering emm_prob
Gene level: no filtering emm_prob
[XClone] multiprocessing for each brk item
nproc: 80
[XClone HMM smoothing] Time used: 14 seconds
[[0.14598567]
[0.66374004]]
[[0.70412148]
[0.24318348]]
[XClone hint] BAF_final_file saved in /storage/yhhuang/users/rthuang/xclone/tutorials/data/.
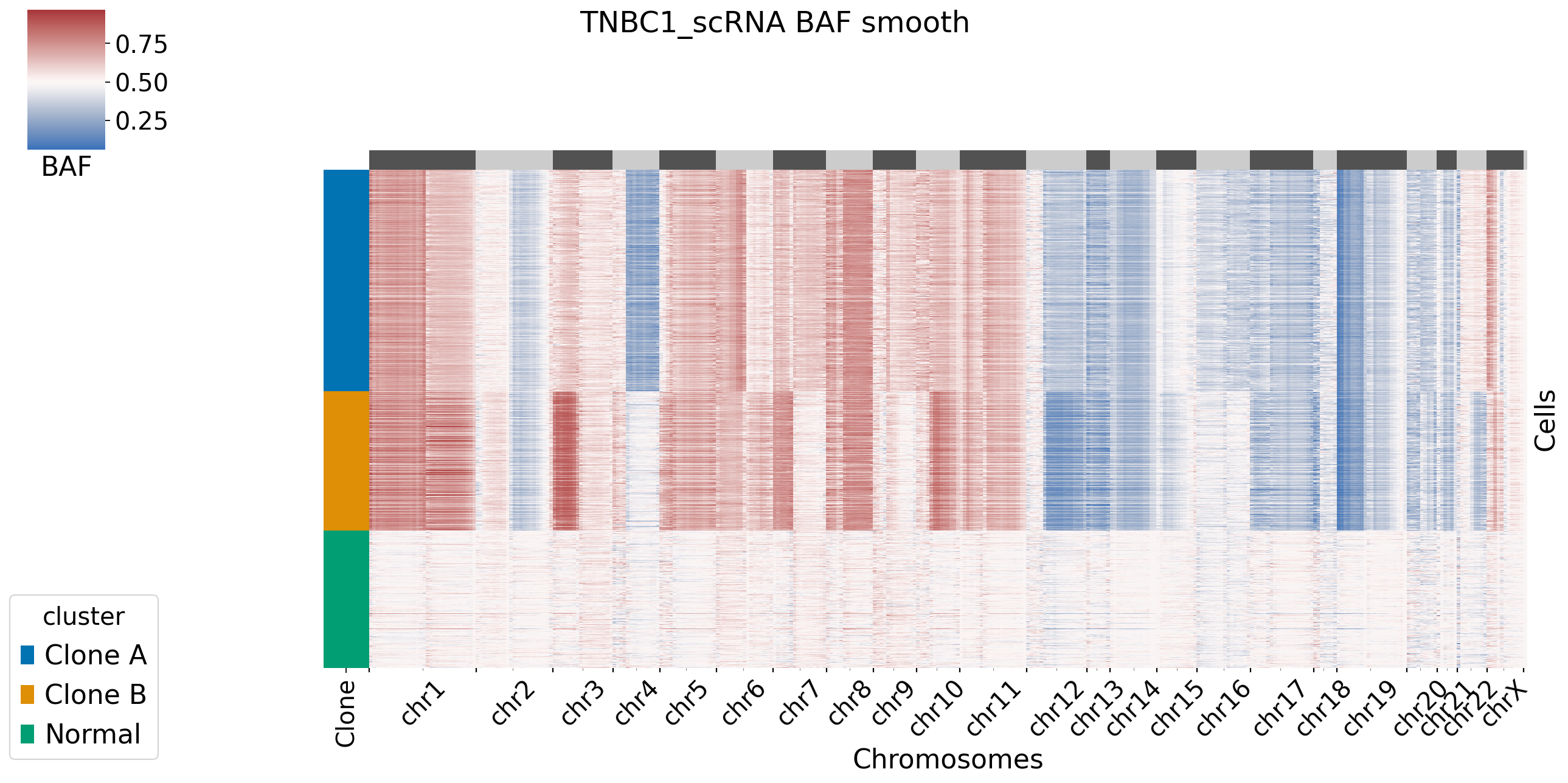
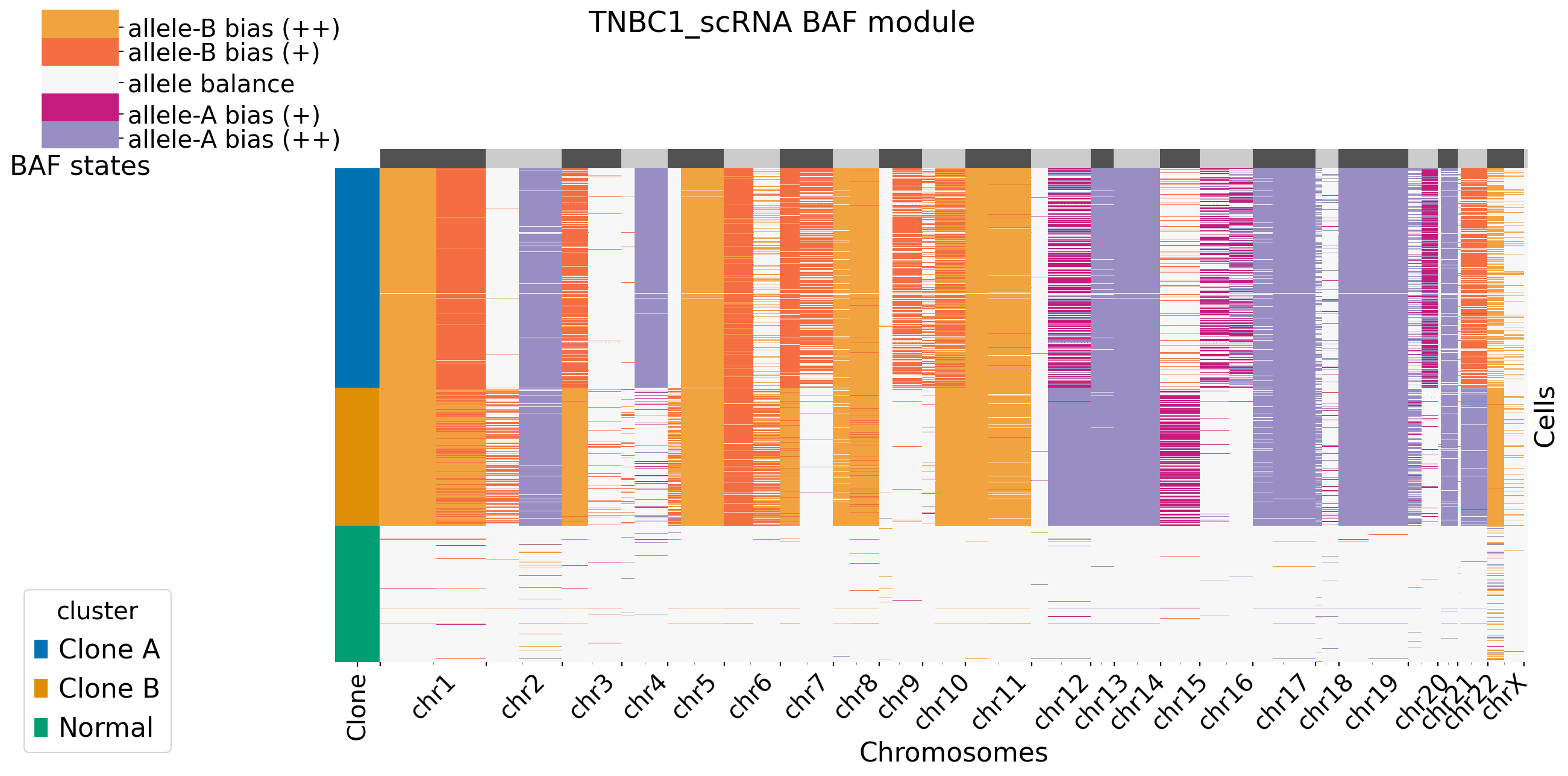
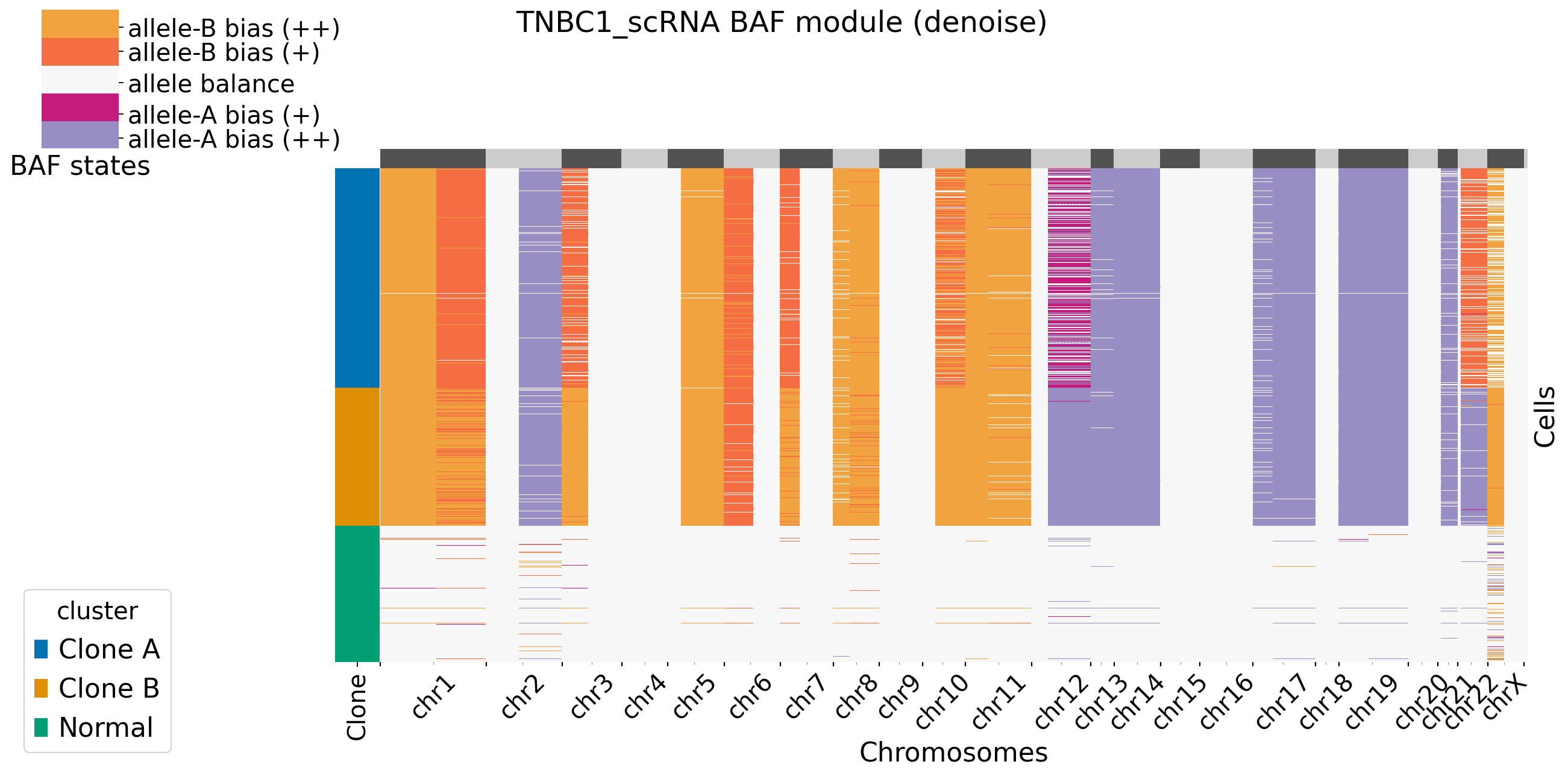
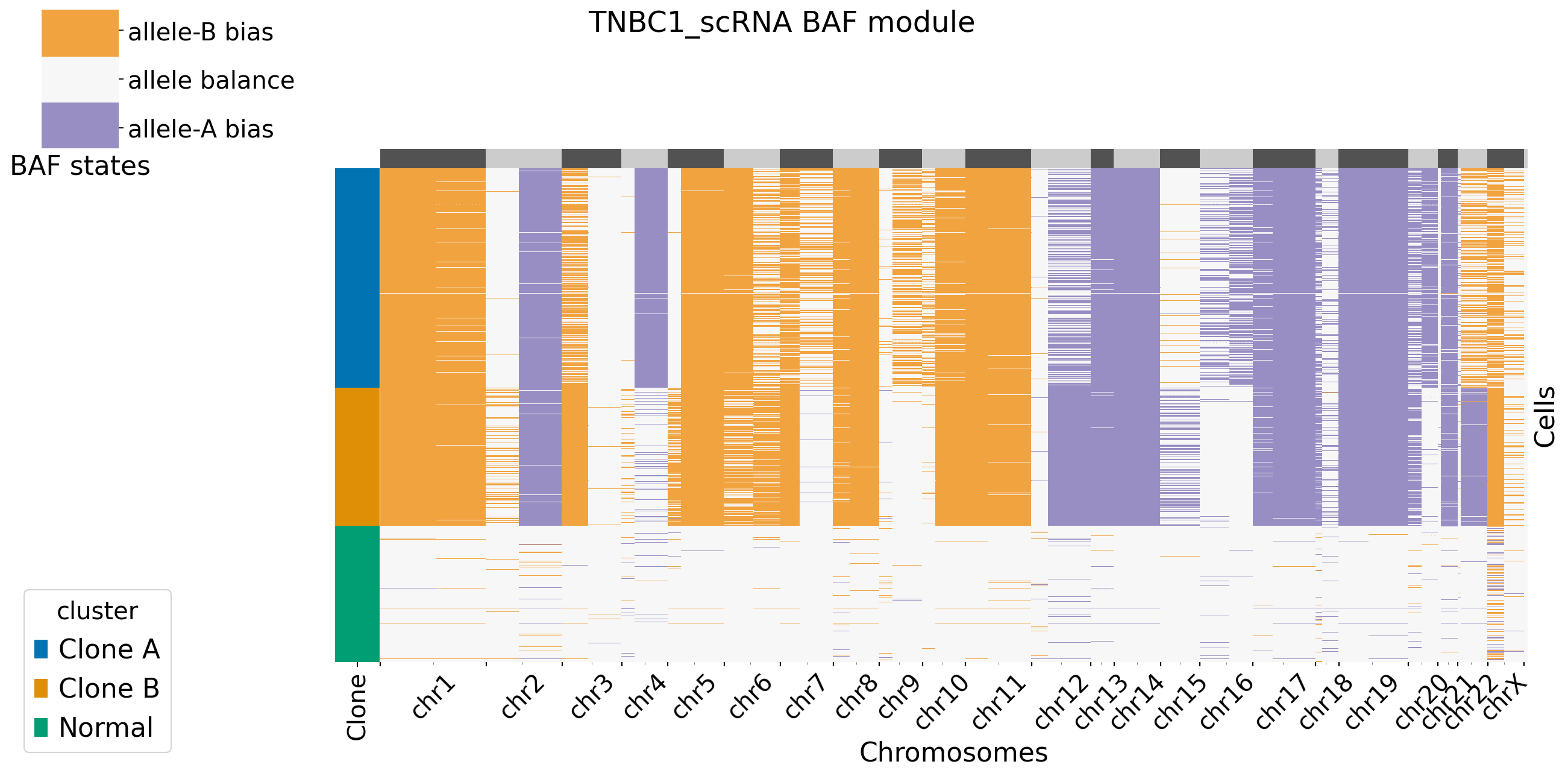
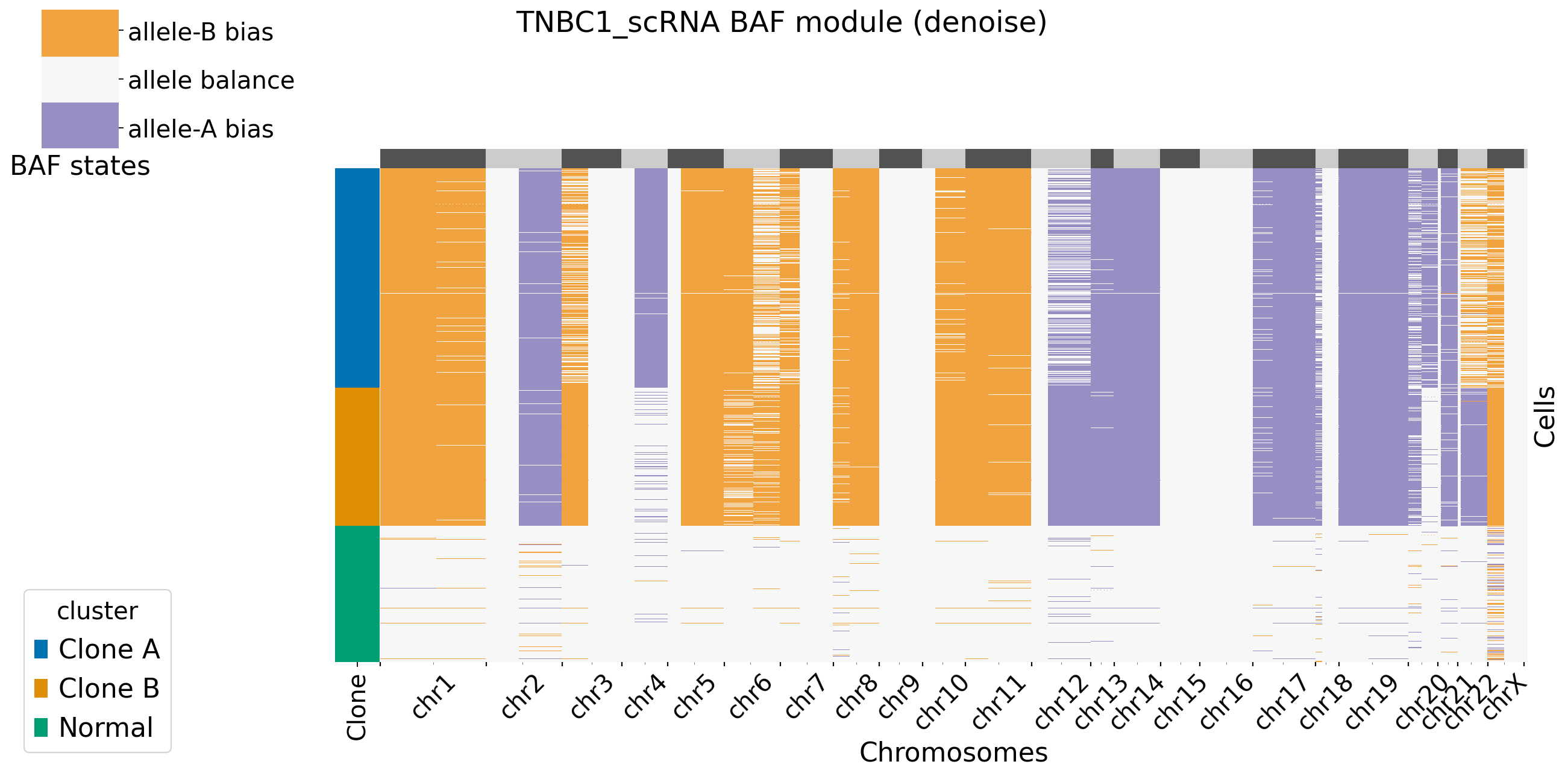
[9]:
BAF_merge_Xdata
[9]:
AnnData object with n_obs × n_vars = 1097 × 347
obs: 'copykat.pred', 'cluster.pred', 'cluster', 'mit_clone_id', 'confident', 'tumor', 'copykat'
var: 'chr', 'start', 'stop', 'arm', 'chr_arm', 'band', 'gene1_stop', 'bin_stop_arm', 'bin_stop_chr_arm', 'bin_stop_band', 'bin_idx', 'bin_idx_cum', 'GeneName_lst', 'GeneID_lst', 'bin_genes_cnt', 'ref_BAF_phased'
uns: 'local_phasing_key', 'local_phasing_len'
layers: 'ad_bin_softcnt', 'ad_bin', 'dp_bin', 'ad_bin_softcnt_phased', 'ad_bin_phased', 'BAF', 'BAF_phased', 'fill_BAF_phased', 'BAF_phased_KNN', 'BAF_phased_KNN_WMA', 'BAF_phased_WMA', 'bin_phased_BAF_specific_center_emm_prob_log', 'bin_phased_BAF_specific_center_emm_prob_log_KNN', 'emm_prob_log_noHMM', 'emm_prob_noHMM', 'posterior_mtx', 'posterior_mtx_log', 'add_posterior_mtx', 'denoised_add_posterior_mtx', 'denoised_posterior_mtx'
obsp: 'connectivities_expr'
varp: 'WMA_connect'
[10]:
BAF_merge_Xdata.layers["posterior_mtx"].shape
[10]:
(1097, 347, 5)
The final step anndata stored in outdir/data/BAF_merge_Xdata_KNN_HMM_post.h5ad and users can get the CNV indentification in layer posterior_mtx.
Before combine, We checked the dimensions of RDR module and BAF module to make sure they are in same dimensions.
[11]:
RDR_Xdata.var
[11]:
| GeneName | GeneID | chr | start | stop | arm | chr_arm | band | ref_avg | dispersion | gene_dispersion_bse | dispersion_capped | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 26 | HES4 | ENSG00000188290 | 1 | 998962 | 1000172 | p | 1p | p36.33 | 0.611296 | 5.409898 | NaN | 2.920260 |
| 27 | ISG15 | ENSG00000187608 | 1 | 1001138 | 1014541 | p | 1p | p36.33 | 6.817276 | 3.253048 | NaN | 2.920260 |
| 38 | TNFRSF4 | ENSG00000186827 | 1 | 1211326 | 1214138 | p | 1p | p36.33 | 0.637874 | 5.979831 | NaN | 2.920260 |
| 39 | SDF4 | ENSG00000078808 | 1 | 1216908 | 1232031 | p | 1p | p36.33 | 1.408638 | 0.223667 | NaN | 0.223667 |
| 43 | UBE2J2 | ENSG00000160087 | 1 | 1253909 | 1273885 | p | 1p | p36.33 | 0.624585 | 0.175076 | NaN | 0.175076 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 33348 | MPP1 | ENSG00000130830 | X | 154778684 | 154821007 | q | Xq | q28 | 1.614618 | 0.779464 | NaN | 0.779464 |
| 33350 | F8 | ENSG00000185010 | X | 154835788 | 155026940 | q | Xq | q28 | 0.554817 | 0.424299 | NaN | 0.424299 |
| 33353 | FUNDC2 | ENSG00000165775 | X | 155025980 | 155060303 | q | Xq | q28 | 0.860465 | 0.115477 | NaN | 0.115477 |
| 33355 | MTCP1 | ENSG00000214827 | X | 155061622 | 155147937 | q | Xq | q28 | 0.873754 | 0.152878 | NaN | 0.152878 |
| 33357 | VBP1 | ENSG00000155959 | X | 155197007 | 155239817 | q | Xq | q28 | 0.750831 | 0.159337 | NaN | 0.159337 |
6402 rows × 12 columns
[12]:
BAF_merge_Xdata.var
[12]:
| chr | start | stop | arm | chr_arm | band | gene1_stop | bin_stop_arm | bin_stop_chr_arm | bin_stop_band | bin_idx | bin_idx_cum | GeneName_lst | GeneID_lst | bin_genes_cnt | ref_BAF_phased | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 29554 | 2530245 | p | 1p | p36.33 | 31109 | p | 1p | p36.32 | 0 | 0 | MIR1302-2HG,FAM138A,OR4F5,AL627309.1,AL627309.... | ENSG00000243485,ENSG00000237613,ENSG0000018609... | 818 | 0.507143 |
| 100 | 1 | 2530064 | 8434838 | p | 1p | p36.32 | 2547460 | p | 1p | p36.23 | 1 | 1 | AL139246.5,TNFRSF14-AS1,TNFRSF14,AL139246.3,FA... | ENSG00000272449,ENSG00000238164,ENSG0000015787... | 870 | 0.517442 |
| 200 | 1 | 8805860 | 13179464 | p | 1p | p36.23 | 8807051 | p | 1p | p36.21 | 2 | 2 | AL357552.2,ENO1,ENO1-AS1,CA6,SLC2A7,SLC2A5,GPR... | ENSG00000228423,ENSG00000074800,ENSG0000023067... | 809 | 0.520401 |
| 300 | 1 | 13196330 | 18486126 | p | 1p | p36.21 | 13201409 | p | 1p | p36.13 | 3 | 3 | PRAMEF13,PRAMEF18,PRAMEF5,PRAMEF8,PRAMEF33,PRA... | ENSG00000279169,ENSG00000279804,ENSG0000027060... | 860 | 0.534310 |
| 400 | 1 | 18631006 | 23838620 | p | 1p | p36.13 | 18748866 | p | 1p | p36.11 | 4 | 4 | PAX7,TAS1R2,AL080251.1,ALDH4A1,IFFO2,UBR4,AL03... | ENSG00000009709,ENSG00000179002,ENSG0000025527... | 756 | 0.553076 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 32996 | X | 109623700 | 128052398 | q | Xq | q23 | 109625172 | q | Xq | q25 | 7 | 342 | KCNE5,ACSL4,TMEM164,AMMECR1,RTL9,CHRDL1,PAK3,C... | ENSG00000176076,ENSG00000068366,ENSG0000015760... | 731 | 0.501661 |
| 33096 | X | 128323620 | 140772679 | q | Xq | q25 | 128600468 | q | Xq | q27.1 | 8 | 343 | AL442647.1,SMARCA1,OCRL,APLN,XPNPEP2,SASH3,ZDH... | ENSG00000225689,ENSG00000102038,ENSG0000012212... | 743 | 0.483389 |
| 33196 | X | 140782405 | 153689010 | q | Xq | q27.1 | 140784871 | q | Xq | q28 | 9 | 344 | CDR1,AL078639.1,AL451048.1,SPANXB1,AC234778.2,... | ENSG00000184258,ENSG00000281508,ENSG0000022926... | 774 | 0.495017 |
| 33296 | X | 153688099 | 156016837 | q | Xq | q28 | 153696593 | q | Xq | q28 | 10 | 345 | SLC6A8,BCAP31,ABCD1,U52111.1,PLXNB3,SRPK3,IDH3... | ENSG00000130821,ENSG00000185825,ENSG0000010198... | 513 | 0.503322 |
| 33372 | Y | 2786855 | 25733388 | p | Yp | p11.2 | 2787699 | q | Yq | q11.23 | 0 | 346 | SRY,RPS4Y1,AC006157.1,ZFY,ZFY-AS1,LINC00278,TG... | ENSG00000184895,ENSG00000129824,ENSG0000027884... | 718 | 0.500000 |
347 rows × 16 columns
[13]:
flag = ~(BAF_merge_Xdata.var["chr"] == "Y")
BAF_merge_Xdata = BAF_merge_Xdata[:, flag]
Combine module
Similarly, by calling the config method xclone.XCloneConfig(), users can create an instance of the XCloneConfig class in the module of “Combine” to set data-specific configures for Combine module.
Set the output directory by specifying outdir.
Users can decide to plot the main figures in each module or not by setting xclone_plot, True or False. If True, can also select cell annotation term plot_cell_anno_key for plotting cells by clusters. If plot_cell_anno_key is not specified, default cell_anno_key= "cell_type" will be used.
For plotting functions in combine moudle, users can set bool variable merge_loss and merge_loh to see if they want to merge allele-specific copy loss/loh states. For more setting in XClone plotting, refer to plotting page. Here in TNBC1 datasets, we merge both the allele-specific copy loss and loh states.
Users can set BAF_denoise = True to combine denoised results from BAF module with RDR module.
Before running the Combine moudle, users can use xconfig.display() to print all configurations used in Combine module for previewing and logging.
Users can run the analysis on your input data by calling the run_combine method by specifying the RDR_Xdata, BAF_merge_Xdata and config_file. If no custmoised config file is used, XClone will load the default config file.
Here shows the example of TNBC1 Combine module.
[14]:
xconfig = xclone.XCloneConfig(dataset_name = dataset_name, module = "Combine")
xconfig.set_figure_params(xclone= True, fontsize = 18)
xconfig.outdir = outdir
xconfig.cell_anno_key = "cluster.pred"
xconfig.ref_celltype = "N"
xconfig.xclone_plot= True
xconfig.plot_cell_anno_key = "cluster"
xconfig.BAF_denoise = True
xconfig.display()
xclone.model.run_combine(RDR_Xdata,
BAF_merge_Xdata,
verbose = True,
run_verbose = True,
config_file = xconfig)
Trying to set attribute `.var` of view, copying.
Combine
Configurations:
BAF_denoise True
KNN_neighbors 10
_file_format_data h5ad
_file_format_figs pdf
_frameon True
_outdir /storage/yhhuang/users/rthuang/xclone/tutorials
_plot_suffix
_start 1680521352.1022782
_vector_friendly True
cell_anno_key cluster.pred
copygain_correct False
copygain_correct_mode None
copyloss_correct True
copyloss_correct_mode 1
dataset_name TNBC1_scRNA
exclude_XY False
file_format_data h5ad
file_format_figs pdf
merge_loh True
merge_loss True
module Combine
outdir /storage/yhhuang/users/rthuang/xclone/tutorials
plot_cell_anno_key cluster
plot_suffix
ref_celltype N
remove_guide_XY False
set_figtitle True
set_smartseq False
warninig_ignore True
xclone_plot True
[XClone Combination module running]************************
[XClone] BAF extend bins to genes.
[XClone data checking]: RDR and BAF in same cell order.
No genes in this bin: 3103 3152 , skip this bin.
No genes in this bin: 21554 21654 , skip this bin.
No genes in this bin: 22754 22839 , skip this bin.
No genes in this bin: 30061 30063 , skip this bin.
[XClone hint] combine_corrected_file saved in /storage/yhhuang/users/rthuang/xclone/tutorials/data/.
[XClone hint] combine_final_file saved in /storage/yhhuang/users/rthuang/xclone/tutorials/data/.
[XClone plotting]
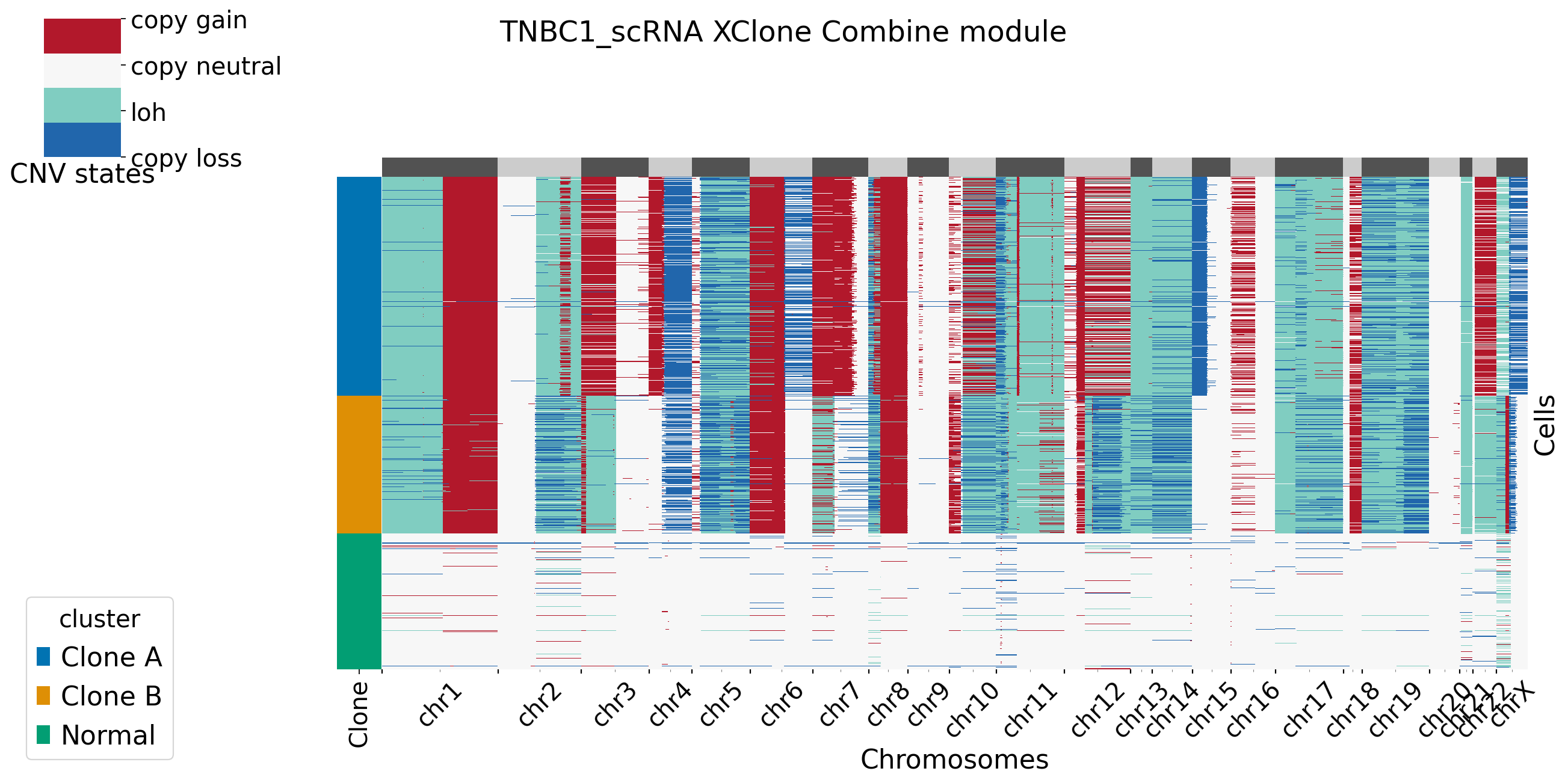
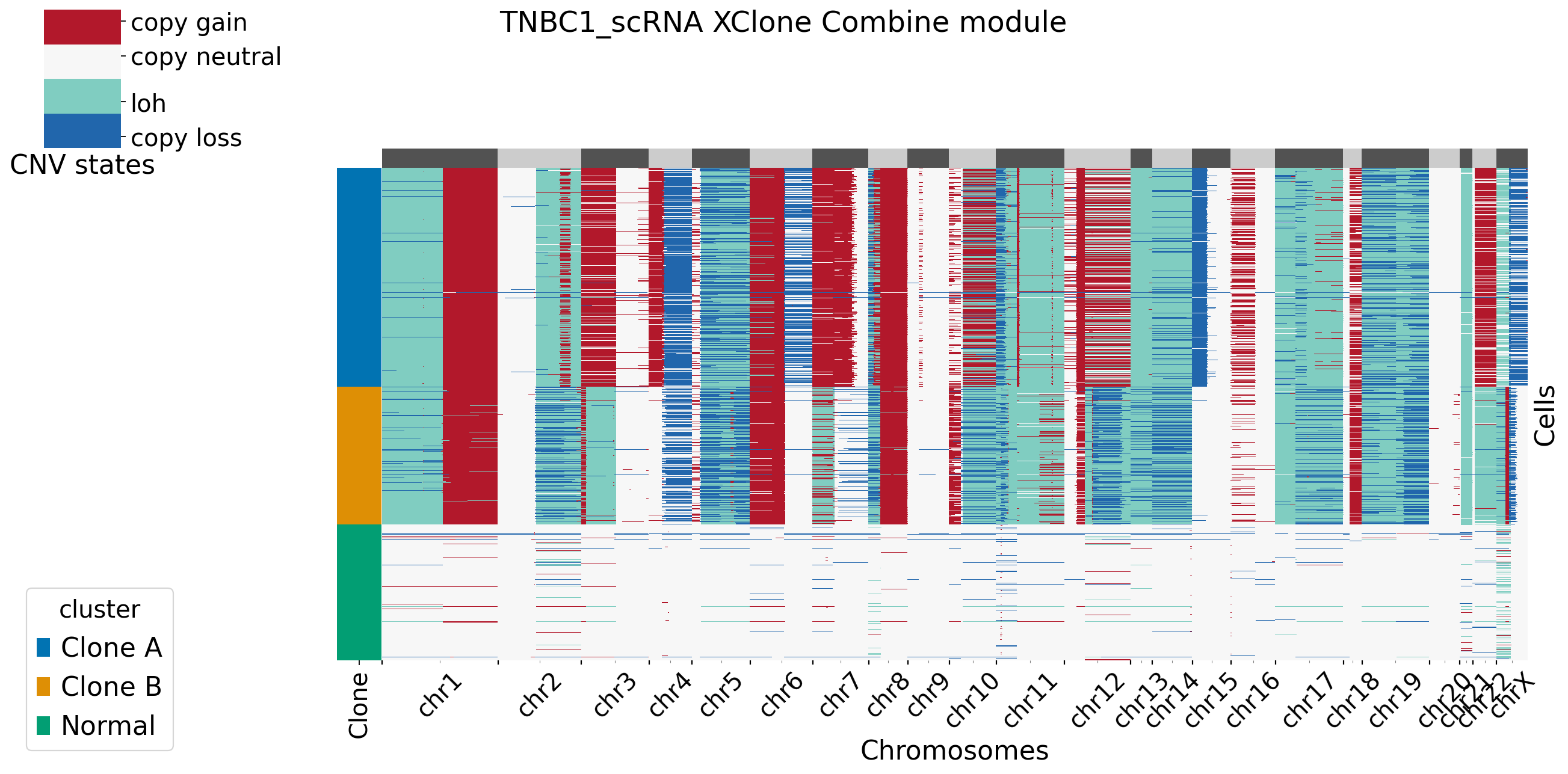
[14]:
AnnData object with n_obs × n_vars = 1097 × 6402
obs: 'copykat.pred', 'cluster.pred', 'cluster', 'mit_clone_id', 'confident', 'tumor', 'copykat', 'counts_ratio'
var: 'GeneName', 'GeneID', 'chr', 'start', 'stop', 'arm', 'chr_arm', 'band', 'ref_avg', 'dispersion', 'gene_dispersion_bse', 'dispersion_capped', 'gene_index'
uns: 'data_mode', 'data_notes', 'genome_mode', 'log', 'rank_marker_genes', 'fit_dispersion_removed_genes', 'dispersion_base_celltype', 'pca', 'neighbors', 'chr_dict', 'guide_CNV_chrs_use_layers', 'guide_CNV_chrs_use_anno_key', 'ref_log_expression_brk', 'group_genes', 'CNV_ratio', 'Logliklihood'
obsm: 'X_pca'
varm: 'PCs'
layers: 'raw_expr', 'raw_ratio', 'ref_normalized', 'expected', 'WMA_smoothed', 'RDR_smooth', 'emm_prob_log', 'emm_prob_log_noHMM', 'emm_prob_noHMM', 'posterior_mtx', 'posterior_mtx_log', 'BAF_extend_post_prob', 'combine_base_prob', 'corrected_prob', 'prob1_merge', 'plot_prob_merge1', 'plot_prob_merge2', 'plot_prob_merge3', 'plot_prob_merge4'
obsp: 'distances', 'connectivities'
The final step anndata stored in outdir/data/combined_final.h5ad and users can get the CNV indentification in layer corrected_prob and prob1_merge.